Lisosomas
Los lisosomas son orgánulos relativamente grandes, formados por el Aparato de Golgi, que contienen enzimas hidrolíticas y proteolíticas encargadas de degradar material intracelular de origen externo (heterofagia) o interno (autofagia) que llegan a ellos. Es decir, se encargan de la digestión celular.
Son estructuras esféricas rodeadas de membrana simple. Son bolsas de enzimas que si se liberasen, destruirían toda la célula. Esto implica que la membrana lisosómica debe estar protegida de estas enzimas. El tamaño de un lisosoma varía entre 0,1-1,2 μm. Los lisosomas fueron descubiertas por el bioquímico belga Christian de Duve en 1974.
En un principio se pensó que los lisosomas serían iguales en todas las células, pero se descubrió que tanto sus dimensiones como su contenido son muy variables. Se encuentran en todas las células animales y vegetales.
Las enzimas lisosomales
El pH en el interior de los lisosomas es de 4,8 (bastante menor que el del citosol, que es neutro) debido a que las enzimas proteolíticas funcionan mejor con un pH ácido. La membrana del lisosoma estabiliza el pH bajo bombeando iones (H+) desde el citosol, asimismo, protege al citosol e igualmente al resto de la célula de las enzimas digestivas que hay en el interior del lisosoma.
Las enzimas lisosomales son capaces de digerir bacterias y otras sustancias que entran en la célula por fagocitosis, u otros procesos de endocitosis.
Los lisosomas utilizan sus enzimas para reciclar los diferentes orgánulos de la célula, englobándolos, digiriéndolos y liberando sus residuos en el citosol. De esta forma, los orgánulos de la célula se están continuamente reponiendo, a través del proceso de digestión de los orgánulos se llama autofagia. Por ejemplo, las células hepáticas se reconstituyen por completo una vez cada dos semanas.
Enzimas más importantes del lisosoma
- Lipasas, que digiere lípidos;
- Glucosidasas, que digiere carbohidratos;
- Proteasas, que digiere proteínas;
- Nucleasas, que digiere ácidos nucleicos.
Formación de lisosomas primarios
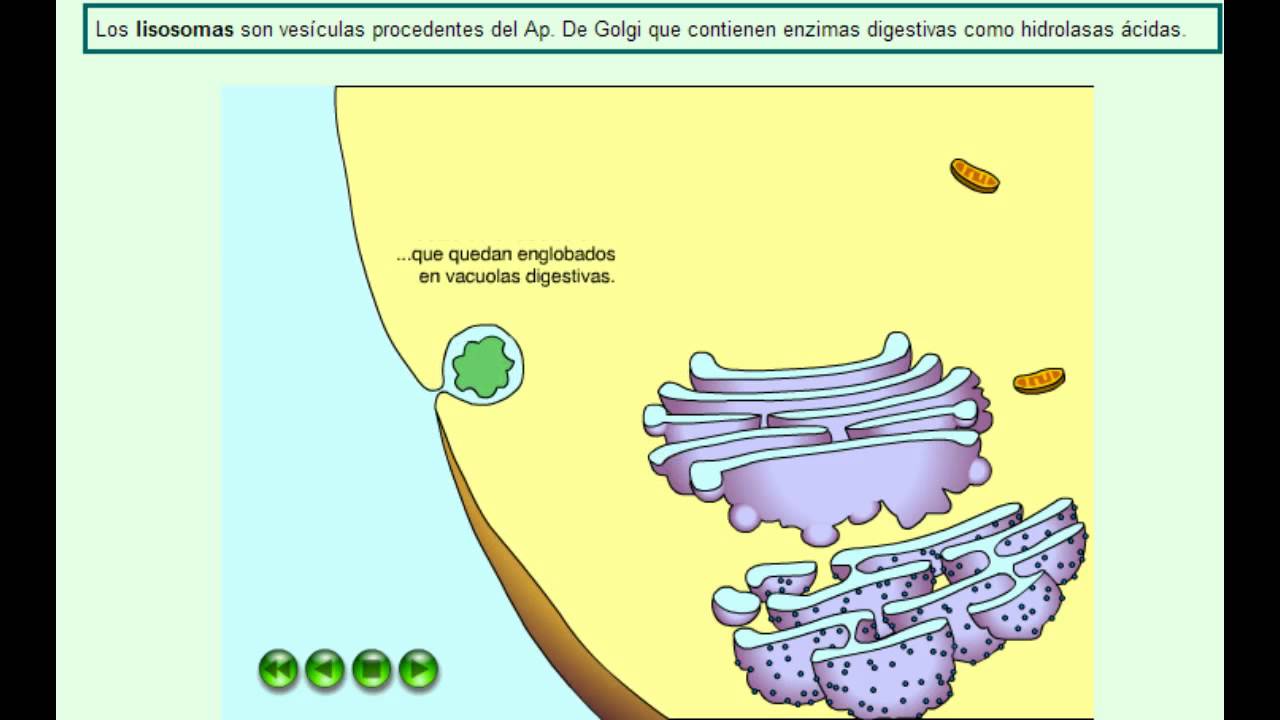
Los lisosomas primarios son orgánulos derivados del sistema de endomembranas. Cada lisosoma primario es una vesícula que brota del aparato de Golgi, con un contenido de enzimas hidrolíticas (hidrolasas). Las hidrolasas son sintetizadas en el retículo endoplasmático rugoso y viajan hasta el aparato de Golgi por transporte vesicular. Allí sufren una glicosilación terminal (proceso químico en el que se adiciona un carbohidrato a otra molécula) de la cual resultan con cadenas glucídicas ricas en manosa-6-fosfato (manosa-6-P). La manosa-6-P es el marcador molecular, la «estampilla» que dirige a las enzimas hacia la ruta de los lisosomas. Se ha estudiado una enfermedad en la cual las hidrolasas no llevan su marcador; las membranas del aparato de Golgi no las reconocen como tales y las empaquetan en vesículas de secreción para ser exocitadas, de modo que, quienes padecen esta enfermedad, acumulan hidrolasas en el medio extracelular, mientras sus células carecen de ellas.
Lisosomas secundarios y digestión celular
Los lisosomas secundarios contienen una variedad de enzimas hidrolíticas capaces de degradar casi todas las moléculas orgánicas. Estas hidrolasas se ponen en contacto con sus sustratos cuando los lisosomas primarios se fusionan con otras vesículas y el producto de la fusión es un lisosoma secundario.
Por lo tanto, la digestión de moléculas orgánicas se lleva a cabo en los lisosomas secundarios, ya que estos contienen a la vez los sustratos y las enzimas capaces de degradarlos.
Existen diversas formas de lisosomas secundarios, según el origen de la vesícula que se fusiona con el lisosoma primario:
- Fagolisosomas: se originan de la fusión del lisosoma primario con una vesícula procedente de la fagocitosis, denominada fagosoma. Se encuentran, por ejemplo, en los glóbulos blancos, capaces de fagocitar partículas extrañas que luego son digeridas por estas células.
- Autofagolisosomas: que son el producto de la fusión entre un lisosoma primario y una vesícula autofágica o autofagosoma. Algunos orgánulos citoplasmáticos son englobados en vesículas, con membranas que provienen de las cisternas del retículo endoplasmático, para luego ser reciclados cuando estas vesículas autofágicas se unen con los lisosomas primarios.
Lo que queda del lisosoma secundario después de la absorción es un cuerpo residual. Los cuerpos residuales contienen desechos no digeribles que en algunos casos se exocitan y en otros no, acumulándose en el citosol a medida que la célula envejece. Un ejemplo de cuerpos residuales son los gránulos de lipofuscina que se observan en células de larga vida, como las neuronas.
Enfermedades lisosómicas
Son enfermedades causadas por la disfunción de alguna enzima lisosómica o por la liberación incontrolada de dichas enzimas en el citosol, lo que produce la lisis de la célula.
En algunos casos, la liberación de las enzimas cumple un papel fisiológico, permitiendo la reabsorción de estructuras que ya no son útiles, por ejemplo la cola de los renacuajosdurante la metamorfosis.
Enfermedades de almacenamiento lisosómico
En las enfermedades de almacenamiento lisosómico, alguna enzima del lisosoma tiene actividad reducida o nula debido a un error genético y el substrato de dicho enzima se acumula y deposita dentro del lisosoma que aumentan de tamaño a causa del material sin digerir, lo cual interfiere con los procesos celulares normales; algunas de estas enfermedades son:
- Esfingolipidosis. Son enfermedades causadas por la disfunción de algunas de las enzimas de la ruta de degradación de los esfingolípidos. Dado que los esfingolípidos abundan en el cerebro, varias de estas enfermedades cursan con retraso mental severo y muerte prematura; entre ellas hay que destacar la enfermedad de Tay-Sachs, la enfermedad de Gaucher, la enfermedad de Niemann-Pick, la enfermedad de Krabbe, la fucosidosis, etc.
- Carencia de lipasa ácida. La lipasa ácida es una enzima fundamental en el metabolismo de los triglicéridos y del colesterol, que se acumulan en los tejidos. La disfunción de esta enzima provoca dos enfermedades, la enfermedad de almacenamiento de ésteres de colesterol, en que la enzima presenta muy poca actividad, y la enfermedad de Wolman, en que la enzima es totalmente inactiva.
- Glucogenosis tipo II o enfermedad de Pompe. Es un defecto de la α(1-4) glucosidasa ácida lisosómica, también denominada maltasa ácida. El glucógeno aparece almacenado en lisosomas. En niños destaca por producir insuficiencia cardíaca al acumularse en el músculo cardíaco causando cardiomegalia, mientras que en adultos el acúmulo es más acusado en músculo esquelético.
- Mucopolisacaridosis. Causadas por la ausencia o el mal funcionamiento de las enzimas necesarias para la degradación moléculas llamadas glicosoaminoglicanos o glucosaminglucanos (antes llamadas mucopolisacáridos).
- Destacan la mucopolisacaridosis tipo I, también conocida como gargolismo o enfermedad de Hurler, en la que existe un defecto de la enzima α-1-iduronidasa, y la mucopolisacaridosis de tipo II o síndrome de Hunter, causada por un error en la enzima iduronato-2-sulfatasa. El —síndrome Sanfilippo—— MPS III, está relacionado con la acumulación de N-heparan Sulfatasa.
Gota
En la gota, el ácido úrico proveniente del catabolismo de las purinas se produce en exceso, lo que provoca la deposición de cristales de urato en las articulaciones. Los cristales son fagocitados por las células y se acumulan en los lisosomas secundarios; estos cristales provocan la ruptura de dichas vacuolas con la consiguiente liberación de enzimas lisosómicos en el citosol que causa la digestión de componentes celulares, la liberación de sustancias de la célula y la autolisis celular.
Artritis reumatoide
La membrana de los lisosomas es impermeable a las enzimas y resistente a la acción de estas. Ambos hechos protegen normalmente a la célula de una batería enzimática que podría degradarla. Existen, sin embargo, algunos procesos patológicos, como la artritis reumatoide, que causan la destrucción de las membranas lisosomales, con la consecuente liberación de las enzimas y la lisis celular.
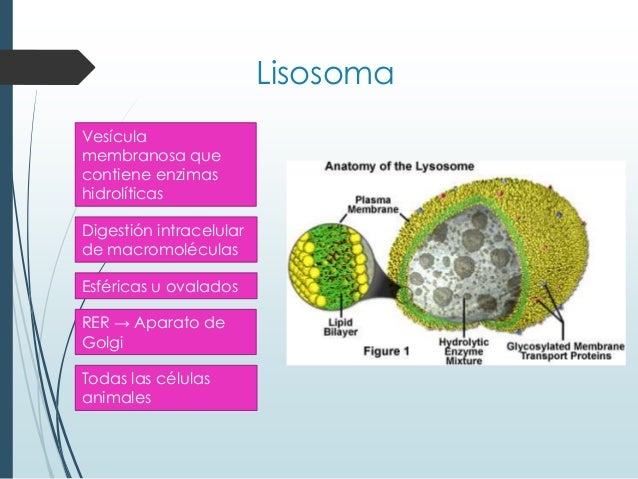
Los lisosomas son organelas formadas por el complejo de Golgi. Contienen enzimas hidroliticas y paleolíticas que se encargan de digerir sustancias externas o internas a ellos, por lo que son los encargados de la digestión celular
Su estructura consiste en una forma redondeada, caracterizadas como una bolsa de enzimas, las cuales en caso de liberarse podrían destruir la célula, por lo que su membrana se encuentra protegida de estas enzimas.
Los lisosomas son propios de células animales, y su tamaño, forma y contenido varían dependiendo de su localización y función.

Función de lisosomas
Las enzimas que contienen los lisosomas son capaces de degradar lípidos, polisacáridos y proteínas, que no van a utilizarse por la célula, por lo que la función de los lisosomas es principalmente degradación de desechos.
Los productos que ya no son útiles para la célula, son transportados a los lisosomas para producir la degradación de estos a moléculas simples, para luego devolverlos al citoplasma y ser reciclados por la célula.
La función primordial de los lisosomas es impedir que sean degradas estructuras necesarias y fundamentales de la célula.
Las enzimas de los lisosomas tienen la capacidad de digerir bacterias y sustancias que ingresan a la célula mediante fagositosis, o endositosis según sea necesario.
Gracias a los lisosomas, las organelas de la célula están en constante reposición y renovación, ya que estos se encargan de degradarlos, proceso llamado autofagia.
Las enzimas más importantes que pueden encontrarse en los lisosomas son, la lipasa, que se encarga de digerir lípidos, glucosidasas para digerir carbohidratos, proteasas, para digerir proteínas, y nucleasas para digerir ácidos nucleicos.


No hay comentarios:
Publicar un comentario